

Germline alterations causing inherited myeloid neoplasia (MN) have become prominent in the 2016 WHO classification. Increasing evidence reveals that many patients with MN (MDS and AML) may indeed harbor unsuspected predisposing or direct driver germline alterations. Penetrance of such alterations may be variable and family history unreliable or absent.
Our previous screening of a selection of genes known to be involved in the pathogenesis of human cancers directly or indirectly according to the difference in the allelic burden in 690 MN patients (Li, S.T, Leukemia, 2020) we identified the myeloperoxidase (MPO) gene as having the highest burden of pathogenic alterations. MPO (17q22-23) encodes a heme-containing enzyme exclusively expressed in myeloid cells. MPO is known for its role in host defense against common microbial infections. In the presence of H2O2, MPO catalyzes the formation of hypohalous acids which are potent oxidants and strong bactericides. Many alterations in MPO have been found in hereditary MPO deficiency, an autosomal recessive syndrome with variable clinical penetrance. MPO deficiency syndrome is characterized by increased susceptibility to infections. To date, formal cancer association has not been established. In particular due to relatively mild phenotype, hereditary deficiency may remain unrecognized.
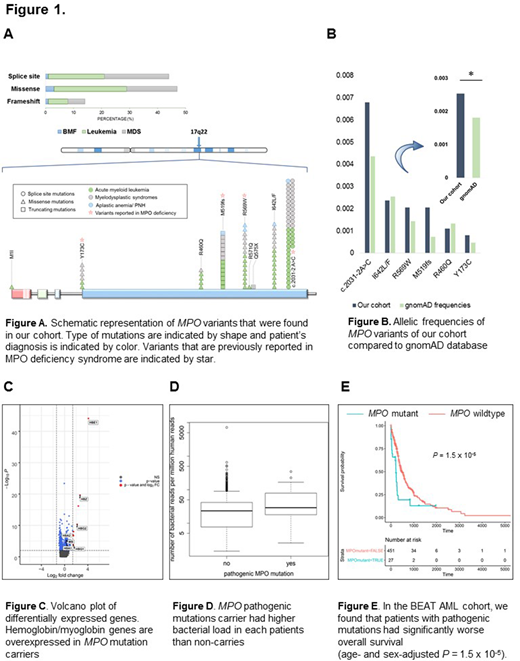
Inspired by results of an initial screen we investigated MPO mutations in 3262 patients with various bone marrow failure syndromes (AA/PNH, 213) and MN (AML, 1655; MDS, 1133; MDS/MPN, 161; MPN, 100). In a subset of patients (20%), paired normal and tumor samples were sequenced in order to confirm germline origin. Germline MPO mutations were identified in 105 cases with 52% of them in AML. The most common recurrent mutation was c.2031-2A>C (42%; 44/105) (3' splice site of intron11) followed by I642L/F 15% (16/105), R569W 14 % (15/105), M519fs 13% (14/105), R460Q 7% (7/105), Y173C 5% (5/105), and R480H 2% (2/105). None of the variants were biallelic. The expected frequency of overall MPO variants in our cohort was higher than gnomAD (0.26% vs. 0.18%; P= .15×10-3) with odds ratio of 1.4. Notably, MPO mutations were significantly enriched in patients with age < 60 (2.2%; odds ratio =12.1, 95%CI=9.3-17.3; P<.0001). Further clinical characterization revealed that the frequencies of MPO mutations were also higher among different disease types than in controls (0.26%, 0.30%, 1.18% for AML, MDS, AA/PNH, P=.008, .001, <.001). Of interest, only the pathogenic variants that had been reported in MPO deficiency syndrome (c.2031-2A>C, R569W, M519fs, Y173C) contributed to this significant enrichment.
Co-occurring lesions of germline MPO mutants (MPOMT) were then investigated: DNMT3A, SF3B1, TP53, NRAS were commonly observed in MPOMT compared to MPO WT-MN (20 vs. 14%, 17 vs. 13%, 16 vs. 8%, and 12 vs. 7%). Normal karyotype was found in 47% of carriers without enrichment for complex, -7/del(7q), del(5q), del(20q) or +8 karyotypes compared to WT. We compared RNA expression patterns between patients with pathogenic MPO mutations and those WT for MPO. Examination of genes significantly overexpressed in MPOMT showed strong enrichment in pseudoperoxidases hemoproteins involved in oxygen transport-globin (P=10-3) and heme biosynthesis (P=.012) (Fig.1C). The overexpression of hemoglobin/myoglobin genes in MPOMT suggests an excessive compensatory process due to the lack of true peroxidase activity. The fact that MPO has microbicidal activity led us to investigate the consequences of MPO mutations. Using shotgun whole-microbiome sequencing of circulating DNA, we examined the overall bacterial load in each patient. We found that MPO pathogenic mutation carriers had significantly higher bacterial burden than non-carriers (Fig.1D). The high bacterial burden of MPO mutants possibly promoted chronic inflammation, known to correlate with cancer pathogenesis. Indeed, we found that the presence of pathogenic MPO mutations was correlated with decreased survival in AML (P = 1.5 x 10-5).
In sum, this is the first cohort showing germline MPO variants as predisposing alterations in MN. MPO variants might cause partial or total MPO deficiency resulting in chronic inflammatory responses by excessive compensation of immunity against invading microbes. This chronic inflammation stages can increase proliferation of HSC or myeloid progenitors and give rise to DNA replication associated mutations.
Maciejewski:Alexion, BMS: Speakers Bureau; Novartis, Roche: Consultancy, Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal